Degradation of mRNA is a fundamental process and knowledge about its mechanisms serves to better understand protein synthesis and gene expression regulation. NMD mechanisms are an essential part of intracellular anti-viral control, shaped gene sequences during evolution, and are required for the development of complex organisms. The mechanistic detail of how RNA to be degraded is recognized and which are the factors involved in the process, remains to be clarified. Preliminary and recently published results allow us to ask new fundamental questions about the mechanisms of NMD (non-sense mediated mRNA decay) and mRNA degradation in general:
- What is the composition of NMD-related protein-RNA complexes?
- How is the degradation system recruited to mRNA substrates?
- How enzymes that degrade NMD substrates are activated?
-
Predict cellular processes sensitive to NMD defects?

NMD is a complex pathway that involves translation termination, the core Upf factors (1, 2, 3) decapping, deadenylation and potentially endonucleolytic cleavage of RNA.
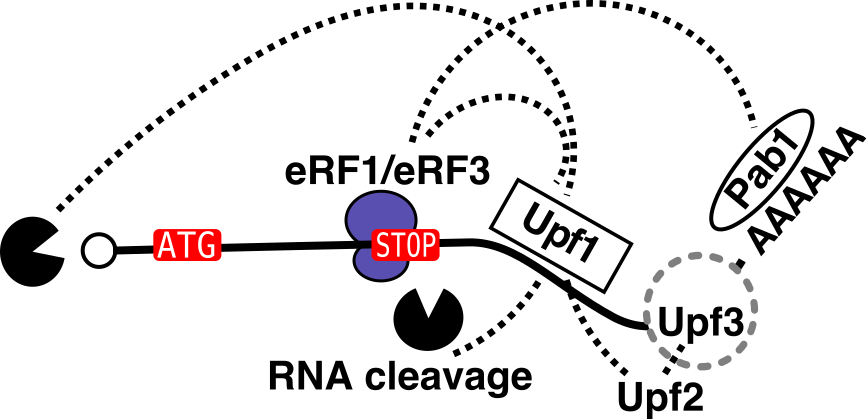
To answer these questions we use affinity purification of RNA-protein and protein-protein complexes coupled with mass-spectrometry based quantitative estimation of protein components and RNASeq for RNA components, large scale genetic screens and classical phenotypic analysis of individual yeast mutant strains.
Our expertise in chemogenomic screens is useful in collaborative studies where the mechanism of action of a toxic compound is sought. Large-scale data analysis and visualization also pushed us to set up interactive graphical views for genetic interactions (PNAS, 2008, NAR, 2021) and protein-protein association results.
Our large-scale protein-protein interaction results on NMD, initially submited to the pre-print server biorxiv (https://www.biorxiv.org/content/early/2018/02/16/266833), are published (EMBOJ, 2018). They show that NMD factors associate in two mutually exclusive complexes around the RNA helicase Upf1. Our results led to the characterization of new direct partners of Upf1 in yeast, the Nmd4 and Ebs1 proteins, that are potential equivalents of human Smg6 and Smg5/7. How these RNA helicase partners affect RNA decay during NMD, what exactly happens on RNA substrates and to what extent these mechanisms are conserved in human cells are questions we are currently addressing.
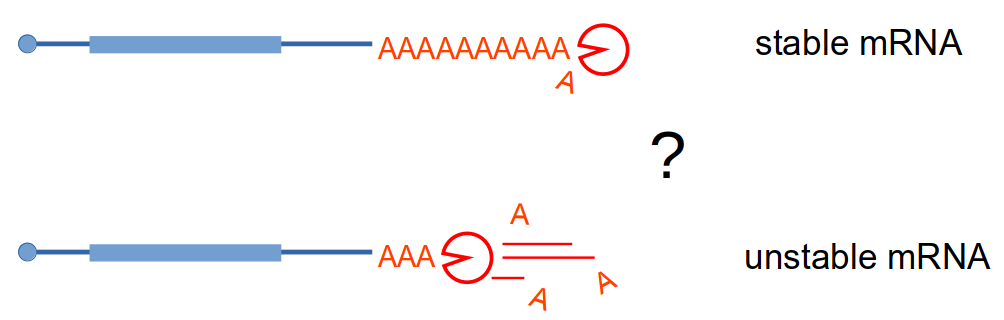 Stimulated by our work on NMD, where deadenylation does not play a role in triggering mRNA degradation, we investigated the question of how the shortening of the poly(A) tail affects mRNA half-life. To our suprise, and contrary to the textbook version of mRNA degradation mechanisms in eukaryotes, we could not identify a global role for deadenylation in mRNA degradation. We used an inducible degron system to rapidly deplete the cell for deadenylation enzymes and Nanopore sequencing to estimate the levels of mRNA and the length of the poly(A) tails. Validating the Nanopore data, reporter mRNA poly(A) tails could be slowly deadenylated without any impact on the transcript half-life. The manuscript describing these results is present on biorxiv.
Stimulated by our work on NMD, where deadenylation does not play a role in triggering mRNA degradation, we investigated the question of how the shortening of the poly(A) tail affects mRNA half-life. To our suprise, and contrary to the textbook version of mRNA degradation mechanisms in eukaryotes, we could not identify a global role for deadenylation in mRNA degradation. We used an inducible degron system to rapidly deplete the cell for deadenylation enzymes and Nanopore sequencing to estimate the levels of mRNA and the length of the poly(A) tails. Validating the Nanopore data, reporter mRNA poly(A) tails could be slowly deadenylated without any impact on the transcript half-life. The manuscript describing these results is present on biorxiv.
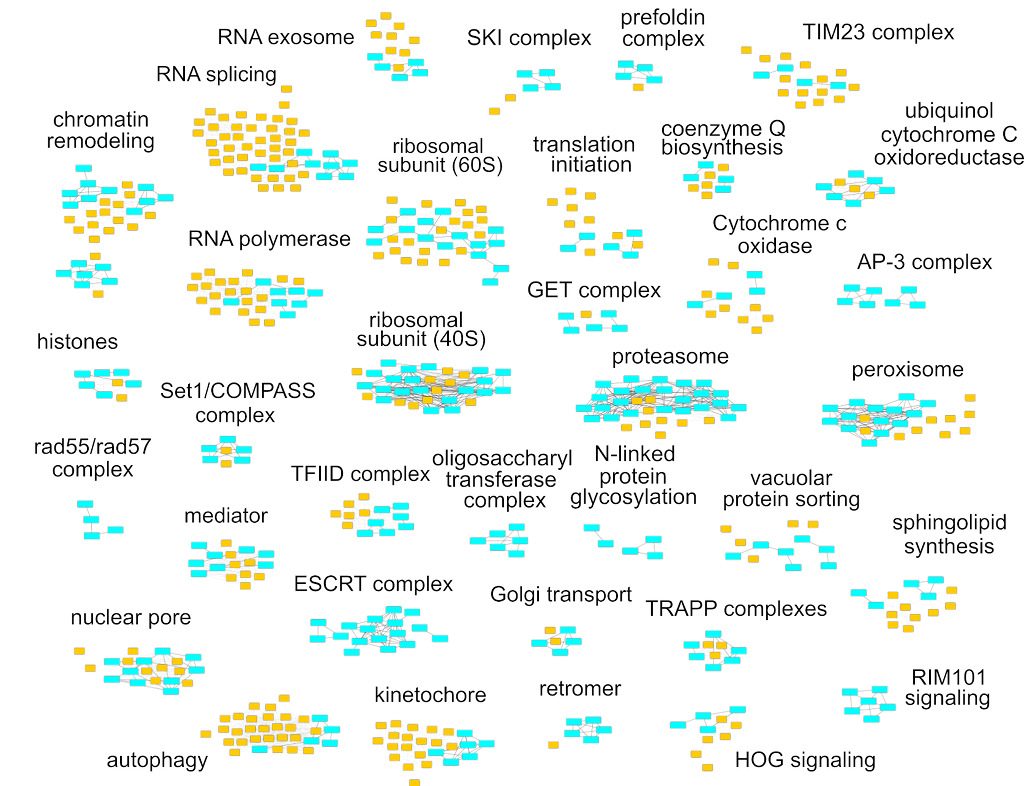
The complexity of RNA metabolism pathways require both biochemical and genetic tools for their investigation. We performed several hundred thousands measures for growth of strains in which two genes were affected. Since genes involved in the same pathway tend to respond similarly to the addition of a second mutation, these genetic interaction screens can be used to identify new factors involved in RNA metabolism. We developed a publication-based method to be able to infer function from genetic interaction profile similarity. An example of the large panel of cellular functions that were explored by our genetic screens in shown below. The genome-wide results, together with validations, showing, for example, the importance of Puf4-dependent post-transcriptional regulation of RPL9B expression, are available (NAR, 2021).
Online resources (interactive):
Genetic interaction profiles and double mutant effects in yeast (700 000 measurements, Nucleic Acids Res, 2021): hub05.hosting.pasteur.fr/GIM_interactions/
Protein interaction network for yeast NMD ( a set of 113 affinity purification and quantitative mass-spectrometry experiments, EMBO J, 2018): http://hub05.hosting.pasteur.fr/NMD_complexes/
The initial data set of genetic interactions obtained by the GIM method (PNAS, 2008): http://hub05.hosting.pasteur.fr/PNAS2008/