La dégradation de l’ARNm est un processus cellulaire qui peut avoir un rôle essentiel dans la régulation de l’expression des gènes. Malgré son importance dans la protection des cellules eucaryotes contre les virus et pour le développement des organismes complexes, les mécanismes moléculaires impliqués restent peu connus. Nos résultats récents ont soulevé des nouvelles questions sur le mécanisme du NMD (dégradation de l’ARNm portant des codons stop prématurés), une des voies majeures de la dégradation des ARN cytoplasmiques chez les eucaryotes. Cette voie est connectée à d’autres mécanismes de dégradation des ARN, qui restent à caractériser :
- Quelle est la composition des complexes ARN-protéine pour la dégradation ?
- Comment les enzymes de dégradation sont-elles recrutées ?
- Comment ces enzymes sont activées ?
- Quels sont les processus cellulaires naturellement affectés par la voie NMD ?

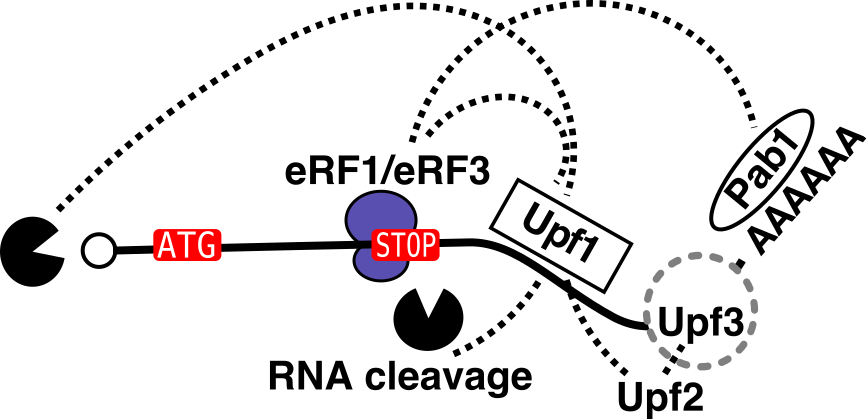
La dégradation par NMD implique la terminaison de la traduction, les facteurs Upf (1, 2, 3) et les enzymes d’enlevement du ‘cap’, de raccourcissement de la queue poly(A) et, potentiellement, de clivage endonucleolytique de l’ARN.
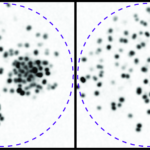
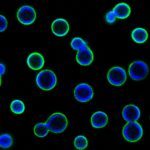
Le travail sur des données à grande échelle nous a permis de développer des nouvelles méthodes d’analyse et de fournir des interfaces de visualisation pour des résultats quantitatifs sur les interactions génétiques et sur les associations protéine-protéine.
Nos résultats sur la dynamique des complexes NMD (EmboJ, 2018) ont défini deux complexes NMD qui ont seulement l’hélicase ARN Upf1 comme composant commun. L’analyse fonctionnelle de Nmd4 et Ebs1, deux facteurs associés physiquement à Upf1 a montré que ces facteurs pouvaient être des équivalents des Smg6 et Smg5-7 des eucaryotes multicellulaires. Ces résultats relancent les études sur le mécanisme du NMD dans l’organisme modèle ou le phénomène a été pour la première fois découvert (avant 1980, laboratoire de François Lacroute).
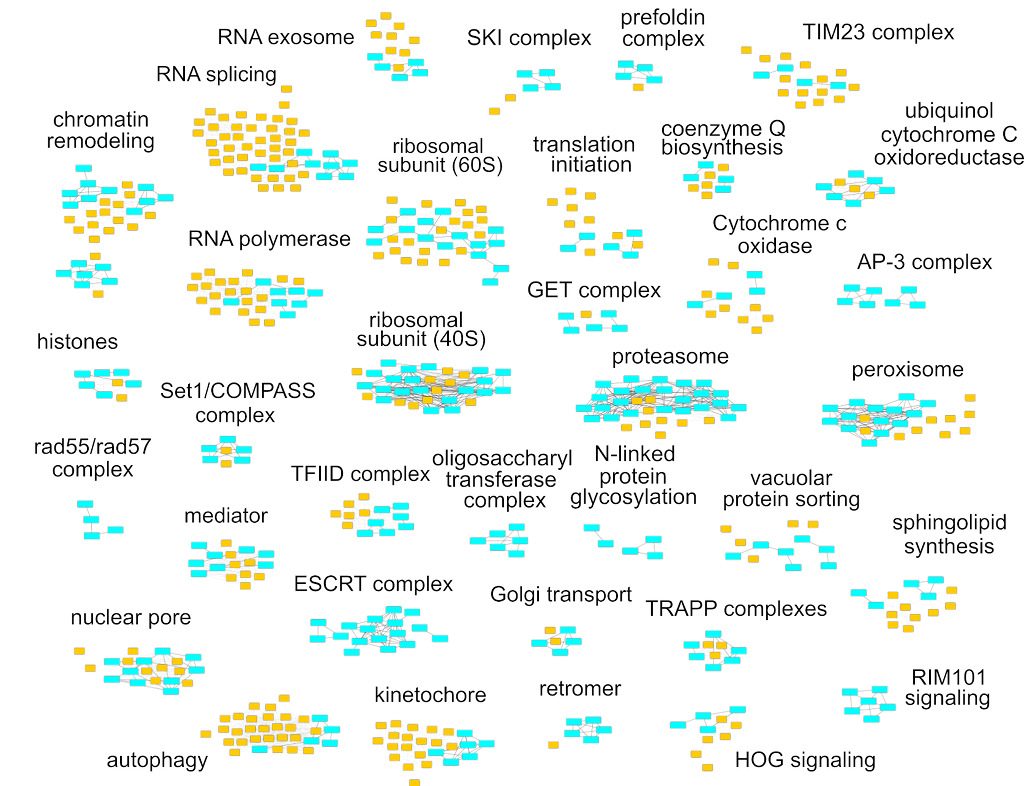
La complexité du métabolisme de l’ARN demande à la fois des outils biochimiques et génétiques pour son exploration. Nous avons obtenu quelques centaines de milliers de mesures de vitesse de croissance pour des souches de levure dans lesquelles deux gènes étaient affectés. Comme les gènes ayant des fonctions similaires ont tendance à répondre de manière similaire au rajout d’une seconde mutation, ces cribles génétiques peuvent servir pour identifier des nouveaux facteurs du métabolisme de l’ARN. Nous avons utilisé les données de la littérature pour développer une méthode nous permettant d’associer des nouveaux gènes à des processus cellulaires connus. Un exemple du panel étendu de fonctions cellulaires que nous avons exploré grâce aux cribles génétiques à grande échelle est montre dans la figure suivante. Le manuscrit qui décrit ce projet a été récemment publié (NAR, 2021).
Accès à nos données (interface web interactive)
Profils d’interactions génétiques pour la découverte de nouveaux liens fonctionnels (700 000 mesures, Nucleic Acids Res, 2021) : hub05.hosting.pasteur.fr/GIM_interactions/
Réseau d’interactions protéiques (set de plus de 100 expériences de purification d’affinité et spectrométrie de masse, EMBO J, 2018) : http://hub05.hosting.pasteur.fr/NMD_complexes/
Le set initial de résultats obtenus par la méthode GIM (PNAS, 2008): http://hub05.hosting.pasteur.fr/PNAS2008/