About
Hydrogen Deuterium eXchange followed by Mass Spectrometry (HDX-MS) is a recognized biophysical tool in structural biology capable of probing protein/ligand interactions, conformational changes, and protein folding and dynamics. The use of improved HDX-MS workflows enables the structural analysis of larger protein systems such as antigen-antibody complexes (> 180 kDa) or integral membrane proteins in a more routine way. The characterization of such biological systems results in very complex HDX-MS datasets, for which specific non-commercial analytical software, as well as commercial platforms, have been developed. However, many of these existing tools do not integrate statistical approaches and use the absolute difference of deuterium uptake to evaluate the significance between conditions.
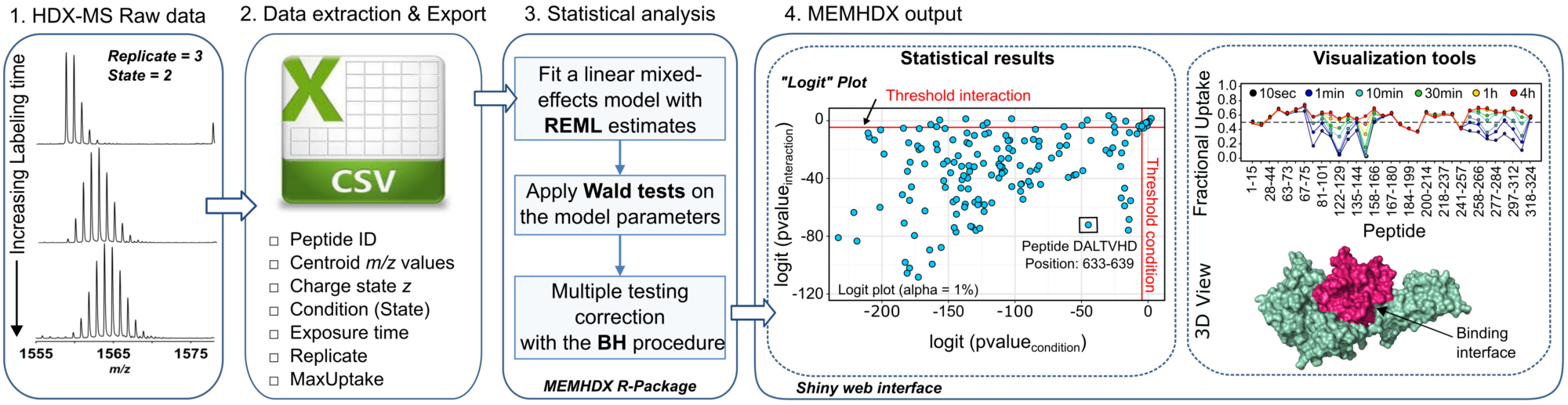
In this regards, we introduced a web application named “MEMHDX” (Mixed-Effects Model for HDX experiments) to aid in the rapid statistical validation and the visualization of large HDX-MS datasets. The initial concept of the software was to complement the Waters HDX-MS platform installed at Institut Pasteur at the end of 2013 (CASCICE Equipex), which is to-date the only complete automated pipeline commercially available. MEMHDX uses a linear mixed-effects model where replicates are considered as random effects. This is a convenient way to account for both the time dependency and the variability across replicates. Moreover, instead of testing the variation in global deuterium exchange between two experimental conditions, we propose to calculate two individual p-values for each peptide. The use of two distinct p-values in the handling of HDX-MS data introduces a novel way to interpret and classify HDX results.