The work of this team to date has focused on the molecular mechanisms underlying auditory function, through the identification of genes responsible for sensorineural deafness in humans. We have brought the cochlea, the sensory organ of mammals, into the molecular era through the genetic dissection we were the first to propose and perform. This strategy aimed to overcome the difficulties encountered in attempts to apply classic biochemical analyses and molecular biology techniques to the very small numbers of cells of each type present in the cochlea. Furthermore, the molecular machineries involved in the diverse functions of cochlear cells consist of components that are generally present in very small amounts. The auditory mechanoelectrical transduction machinery of the sensory cells, the so-called “hair cells” converts the mechanical sensory signal into an electrical signal.
The genetic approach, the efficacy of which is not dependent on the number of cells expressing the gene of interest, or the resulting number of molecules encoded in each cell, has proved highly fruitful. As soon as the first genes responsible for sensorineural deafness were identified in humans, it became possible, thanks to the biophysicists, physiologists, cell biologists and biochemists who subsequently joined the laboratory, to identify the functions of the proteins encoded by these so-called “deafness” (technically a misnomer as these genes encode proteins with genuine functions and it is only their defects that lead to deafness) genes, by obtaining and integrating a set of complementary data. The mouse models of human forms of deafness generated as a result (through constitutive or conditional gene inactivations) made it possible to determine the embryonic, postnatal, or even stress-associated (particularly for sound stress) role of any given protein. By grouping the proteins encoded by these genes together into protein complexes or networks, we were able to shed light on the molecular mechanisms underlying diverse cochlear functions.
The collaborative studies performed with Professor Paul Avan to elucidate the roles of the proteins encoded by the genes responsible for early-onset deafness led to the discovery of unsuspected roles for several cochlear structures. Diverse fibrous links were identified in the hair bundle, the sound antenna of the sensory cells and the site of auditory transduction. Thus, for example, in children, studies of DFNB16 (the most frequent form of deafness after DFNB1, caused by a defect of the GJB2 gene encoding connexin 26) led to the discovery of a protein, stereocilin, which makes up the fibrous links in the hair bundles of the outer hair cells (figure 1). This work revealed that these links played an essential role in the cooperative opening of the auditory mechanoelectrical transduction channels. In parallel, we revealed the pathogenesis of a very large number of deafness forms, which enabled us to propose a pathophysiological classification. These advances formed a cornerstone of knowledge indispensable for research into curative treatments for certain forms of deafness to become a realistic objective. Our team has, for the last few years, been engaged in the development of gene therapies targeting hereditary early-onset forms of deafness, alongside the team of Dr. Saaid Safieddine (see the project of Dr. Saaid Safieddine). Only our new projects are presented below.
 Hair bundles from the outer hair cells of wild-type mice (Strc+/+) are shown on the left, with hair bundles from the outer hair cells of stereocilin-deficient mice (Strc-/-) on the right. The black arrow indicates the fibrous apical links of the outer hair cells, which disappear in the absence of stereocilin, and the black arrowhead shows the unique fibrous link associated with the mechanoelectrical transduction machinery.
Hair bundles from the outer hair cells of wild-type mice (Strc+/+) are shown on the left, with hair bundles from the outer hair cells of stereocilin-deficient mice (Strc-/-) on the right. The black arrow indicates the fibrous apical links of the outer hair cells, which disappear in the absence of stereocilin, and the black arrowhead shows the unique fibrous link associated with the mechanoelectrical transduction machinery.
Verpy, E., Weil, D., Leibovici, M. et al. Stereocilin-deficient mice reveal the origin of cochlear waveform distortions. Nature 456, 255–258 (2008). https://doi.org/10.1038/nature07380
BASIC HEARING MECHANISMS
We have adopted two complementary approaches, to decipher the complexes and molecular networks involved in the major functions of auditory sensory cells, and more specifically, auditory mechanoelectrical transduction (and the generation of sound distortions that accompanies it), and the synaptic exocytosis of a neurotransmitter (glutamate).
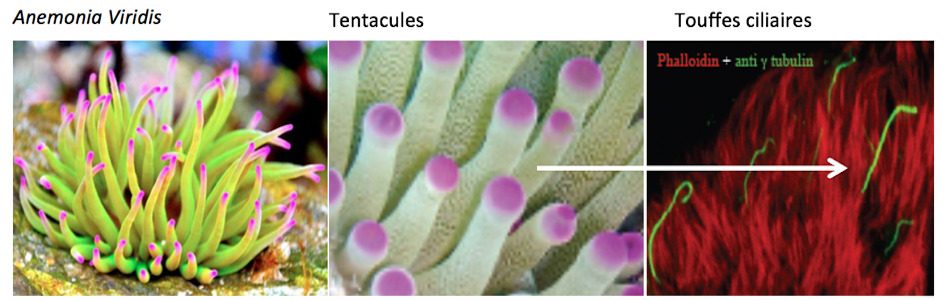
The first of these approaches, performed in collaboration with the laboratory of Eric Röttinger from the IRCAN (l’Institut de Recherche sur le Cancer, the Cancer Research Institute, Inserm U1081-CNRS UMR7284) in Nice, makes use of the conservation of certain functions, including mechanotransduction, in the sea anemone, Nematostella vectensis, a cnidarian invertebrate (figure 2). The second approach, in collaboration with Dr Elise Pepermans from the Center for Proteomics of the University of Antwerp, directed by Geert Baggerman), is designed to decipher the molecular complexes involved in the abovementioned functions in mice, through time-of-flight mass spectrometry, which has a very high resolution (sensitivity theoretically in the attomolar range). These two approaches will also contribute to the identification of genes involved in human deafness (see below).
The molecular machinery responsible for mechanoelectrical transduction in the sea anemone
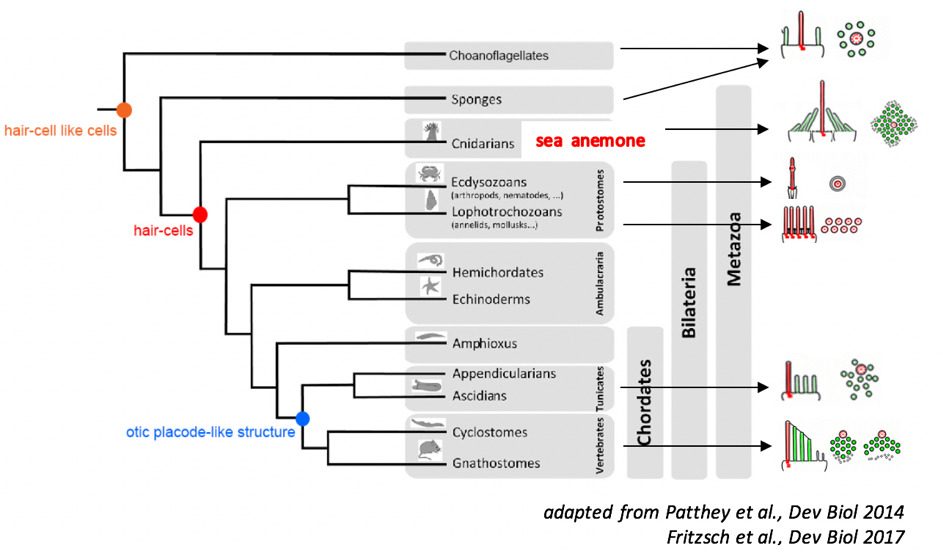
This work is based on the hypothesis that, during the course of evolution, an ancestral molecular machinery for mechanotransduction gave rise to both the mechanoelectrical transduction system common to the hair cells of the auditory sensory organ and the vestibular cells of the balance organ, and that present in certain neurons of the tentacles of sea anemones (figure 2). In vertebrates, the hair cells bear a hair bundle at their apex, which is bathed in a fluid, the endolymph. The hair bundle detects movements of this liquid provoked by sound waves within the cochlea and acceleration in the vestibule.
In sea anemones, the hair bundle at the apex of sensory neurons detects waves in the fluid created by nearby prey and predators.In support of this hypothesis of homology, the morphology of the hair bundles of the sensory neurons of sea anemones is similar to that of the hair bundles in mammalian hair cells, certain transduction physiology features are common to these two structures, and some of the molecular components of these two machineries appear to be shared. From a fine understanding of transduction at the heart of the hair bundle in sea anemones, at the molecular scale, we should be able to find a way to gain effective access to that operating in the hair cells of mammals. Sea anemones are easy to grow in culture, which should make it possible to use classical biochemical approaches, the sequence of their genome has been established and they are genetically modifiable.
With this model, we should be able to reduce the need to use vertebrates, and rodents in particular, as molecular models for obtaining knowledge in the domain of hearing.
The molecular machineries underlying the principal functions of the auditory sensory cells of the cochlea
Our knowledge of these machineries and sensory cells remains partial. It follows that we still know nothing, for example, about the mode of ion channel stimulation in auditory mechanoelectrical transduction. The most recent generation of mass spectrometers has a much higher sensitivity than those that preceded it. A biochemical approach to the protein components of the abovementioned machineries, can, therefore, now be envisaged. This should make it possible to identify the ubiquitous proteins, the posttranslational modifications of the machinery and modulations of the composition of its complexes/networks under different environment or genetic conditions.
PRECISION MEDICINE
By definition, precision (or personalized) medicine makes use of a set of biological, environmental or social data specific to the patient to obtain a better assessment of the patient’s risk of developing disease, to refine diagnosis and to personalize preventive and curative treatments.
Our objective is to develop precision medicine in two domains: the diagnosis of hearing loss and the development of curative treatments for hearing loss. In both cases, we will focus principally on forms of deafness affecting adults, including presbycusis in particular.
We will follow three lines of research:
- Identification of the genes responsible for presbycusis and of the genes and factors conferring susceptibility/resistance to noise-induced hearing loss in humans.
- Development of innovative multiparametric diagnostic approaches for hearing loss.
- Treatment of late-onset deafness by gene therapy: certain treatments for early-onset deafness will also be studied in collaboration with the group of Dr. Saaid Safieddine.
The search for the genes responsible for presbycusis and susceptibility to noise-related hearing loss
Hearing loss is dominated by presbycusis, a form of hearing loss linked to age, which is by far the most frequent form of sensorineural deafness. According to the WHO, 320 million people aged 65 years or over, around the world, currently suffer from presbycusis. This condition leads to social isolation and desocialization, affecting quality of life, in particular. Affected individuals are at risk of depression and cognitive decline, leading to a progressive loss of autonomy. Recent results have suggested that hearing loss during and after middle age is the principal modifiable risk factor for dementia. Like other common diseases and disorders, presbycusis is a multifactorial condition with both genetic and environmental components. The principal environmental factor is overexposure to noise, which is currently the leading aggressor of the auditory system.
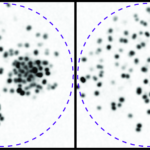
With a view to improving our understanding of the pathophysiology of presbycusis, several laboratories are currently searching for genetic predisposition factors. Efforts to decipher the genetics of presbycusis have advanced considerably in recent years, thanks to the development of genome-wide association studies (GWAS) on patient cohorts of ever-increasing size. A number of genomic variants of genes that could potentially confer a predisposition to the development of presbycusis have been identified. Each make only a very modest contribution to the risk of presbycusis. We have decided to adopt a different approach, by searching for monogenic forms of presbycusis that could potentially be treated by gene therapy and could also directly shed light on the pathophysiology of this condition. Our preliminary results, obtained in a nationwide multicenter study in France, are very encouraging (figure 3). They show that the genes responsible for dominant forms of early-onset deafness are also responsible for severe forms of presbycusis.

The principal objective of the search for human genes responsible for susceptibility/resistance to noise or encoding factors underlying predisposing factors is to decipher the molecular networks and signaling pathways involved, with a view to orienting the search for treatments that protect or repair damaged cells, alongside the development of gene therapy. This work will be performed in the framework of a consortium.
Development of multiparametric diagnosis for hearing loss
The project to develop multiparametric diagnosis for hearing loss aims to refine the diagnosis of hearing loss to make it possible to propose appropriate treatments. Its other objective will be to establish conditions for optimizing tests on candidate drugs. This will make it possible to optimize clinical trials and to define the “profiles” of responders to the diverse treatments to come. We plan to develop a fine, multiparameter description of late-onset forms of human deafness. We will therefore need to obtain and integrate psychophysical, physiological and molecular data. The CERIAH, directed by Professor Paul Avan (see below), with the team of Professor Hung Thai-Van, will develop the necessary methodological and instrumental innovations to achieve this goal and the “Basic hearing mechanisms and precision medicine” team will develop innovative molecular tests.
Treating late-onset deafness
We are considering developing gene therapy approaches.