Lien vers Pubmed [PMID] – 30602788
Lien DOI – 10.1038/s41586-018-0818-3
Nature 2019 01; 565(7738): 230-233
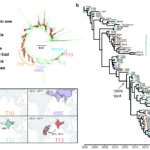
Yemen is currently experiencing, to our knowledge, the largest cholera epidemic in recent history. The first cases were declared in September 2016, and over 1.1 million cases and 2,300 deaths have since been reported1. Here we investigate the phylogenetic relationships, pathogenesis and determinants of antimicrobial resistance by sequencing the genomes of Vibrio cholerae isolates from the epidemic in Yemen and recent isolates from neighbouring regions. These 116 genomic sequences were placed within the phylogenetic context of a global collection of 1,087 isolates of the seventh pandemic V. cholerae serogroups O1 and O139 biotype El Tor2-4. We show that the isolates from Yemen that were collected during the two epidemiological waves of the epidemic1-the first between 28 September 2016 and 23 April 2017 (25,839 suspected cases) and the second beginning on 24 April 2017 (more than 1 million suspected cases)-are V. cholerae serotype Ogawa isolates from a single sublineage of the seventh pandemic V. cholerae O1 El Tor (7PET) lineage. Using genomic approaches, we link the epidemic in Yemen to global radiations of pandemic V. cholerae and show that this sublineage originated from South Asia and that it caused outbreaks in East Africa before appearing in Yemen. Furthermore, we show that the isolates from Yemen are susceptible to several antibiotics that are commonly used to treat cholera and to polymyxin B, resistance to which is used as a marker of the El Tor biotype.