Link to Pubmed [PMID] – 31127303
Mol. Biol. Evol. 2019 Sep;36(9):2069-2085
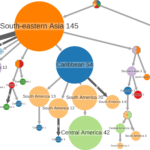
The reconstruction of ancestral scenarios is widely used to study the evolution of characters along phylogenetic trees. One commonly uses the marginal posterior probabilities of the character states, or the joint reconstruction of the most likely scenario. However, marginal reconstructions provide users with state probabilities, which are difficult to interpret and visualize, whereas joint reconstructions select a unique state for every tree node and thus do not reflect the uncertainty of inferences. We propose a simple and fast approach, which is in between these two extremes. We use decision-theory concepts (namely, the Brier score) to associate each node in the tree to a set of likely states. A unique state is predicted in tree regions with low uncertainty, whereas several states are predicted in uncertain regions, typically around the tree root. To visualize the results, we cluster the neighboring nodes associated with the same states and use graph visualization tools. The method is implemented in the PastML program and web server. The results on simulated data demonstrate the accuracy and robustness of the approach. PastML was applied to the phylogeography of Dengue serotype 2 (DENV2), and the evolution of drug resistances in a large HIV data set. These analyses took a few minutes and provided convincing results. PastML retrieved the main transmission routes of human DENV2 and showed the uncertainty of the human-sylvatic DENV2 geographic origin. With HIV, the results show that resistance mutations mostly emerge independently under treatment pressure, but resistance clusters are found, corresponding to transmissions among untreated patients.