


About
In order to understand the spread of infectious diseases, we are often interested in the transmission relationship between pairs of cases. The use of pathogen sequences has revolutionized our ability to characterize the relationship between cases – by looking at the genetic sequence of the pathogen that made different individuals sick, we can reconstruct the spread of pathogens and identify factors associated with spread.
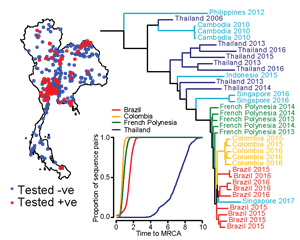
The Mathematical Modelling of Infectious Diseases Unit works in developing mathematical models that uses pathogen sequence data to better understand the epidemiology of different pathogens. In collaboration with Thai Ministry of Public Health, we have used Zika sequences to demonstrate that Zika virus has silently circulated in Thailand for 16+ years. This has clear implications for the ongoing surveillance for severe complications associated with Zika infected.
The use of sequences is particularly useful for endemic pathogens, where the co-circulation of many different overlapping transmission chains can effectively hide the underlying patterns of spread. By sequencing thousands of dengue viruses from Thailand (where dengue has circulated for decades) in collaboration with the University of Florida, WRAIR and partners in Thailand, we have demonstrated that it takes one year for a viral strain to be well mixed throughout the country but that there is limited international spread. We are employing similar approaches with Bordetella Pertussis using full genome sequences from throughout Europe and over a 25 year time period, in collaboration with the French National Reference Center for Pertussis, based at Institut Pasteur (headed by Dr. Sylvain Brisse).