






About
Cardiac malformation is a major public health issue, yet its origin remains poorly understood. It is the most frequent birth defect (1%), the main cause of mortality before the first year of life and often requires lifelong follow-up. Collectively, congenital heart defects (CHD) are common, but individually they may correspond to rare disorders, requiring expert diagnosis and sophisticated repair surgery. Despite their burden, CHD have a known genetic origin in about 30% of cases, mainly cardiomyopathies and syndromes. However, the origin of structural heart defects remains elusive.
The shape of the heart conditions its function, in two parallel but asymmetric left/right circulations. Circulations involve analogous cardiac compartments (vein-atrium-ventricle-arterial trunk), but with a specific left/right anatomy, adapted to high/low pressure and connected to specific vascular networks. Structural heart defects may affect the size, connection or septation of cardiac compartments. They alter blood circulation or oxygenation, leading to heart dysfunction. Pathological mechanisms cannot be studied in patients, because they occur during embryonic development. The mouse provides the best experimental model to study the origin of structural congenital heart defects.
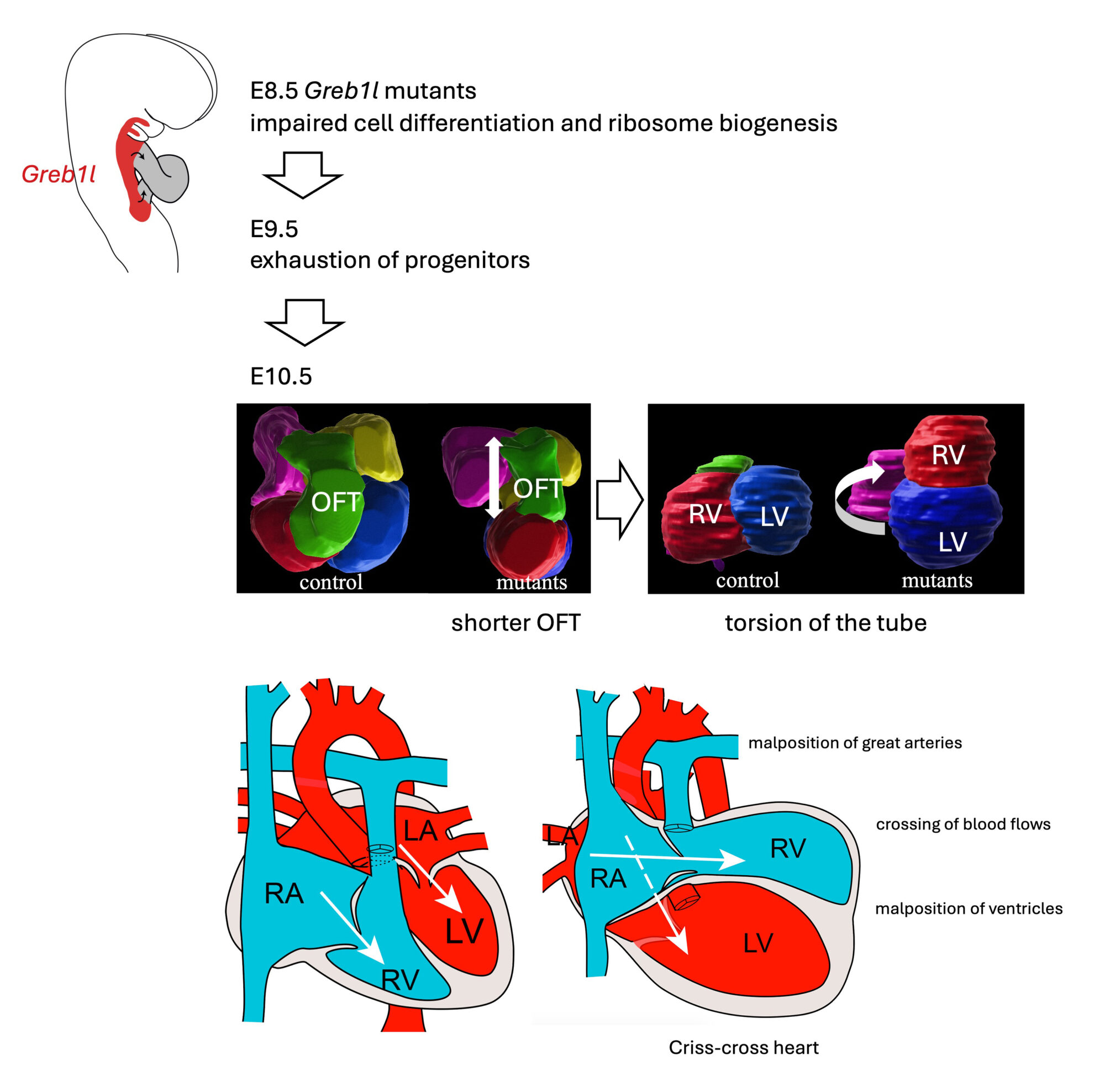
GREB1L is associated with a spectrum of congenital defects in humans (renal agenesis, hearing impairment, thymic, genital, and skeletal anomalies), yet its function was poorly understood. In mice and cardioids, we uncovered its role in maintaining a reservoir of precursor cells. Inactivation of Greb1l in the mouse impairs the elongation of the heart tube leading to criss-cross heart, with twisted atrio-ventricular connections, supero-inferior ventricles and malposition of the great arteries. We thus identified the first mouse model of criss-cross heart and deciphered the pathological mechanism of this rare structural congenital heart defect (1/125,000 birth).
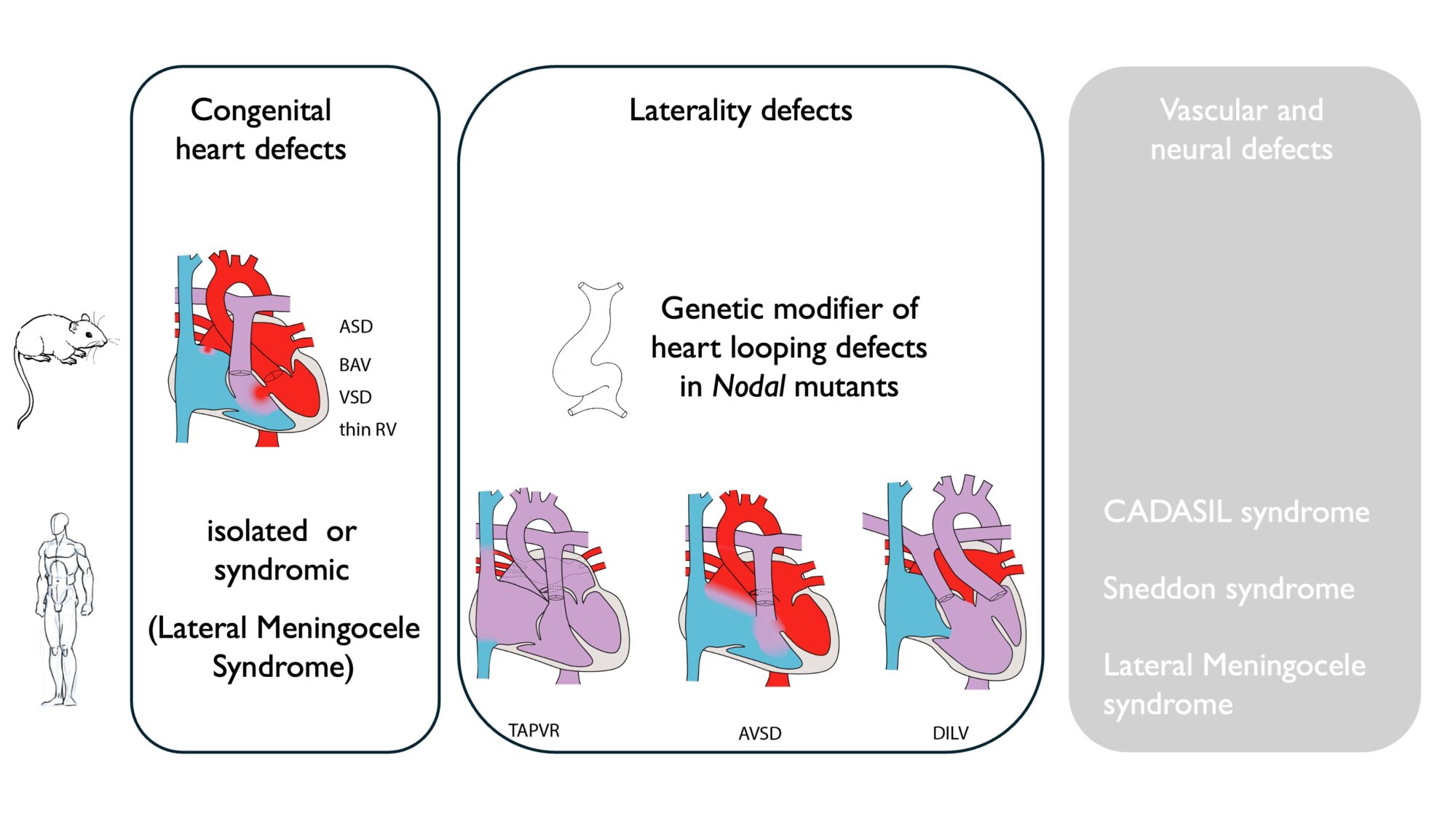
We have identified Notch3 as a novel asymmetric gene. In single mutants, we observe that Notch3 alone is required with partial penetrance for ventricle thickness, septation and aortic valve, in addition to its known role in coronary artery smooth muscle, but not for embryonic heart looping. In compound mutants, we show that Notch3 does not interact genetically with Nodal, but its absence modifies the phenotype of Nodal mutants, modulating looping direction and the curvature of the outflow tract. Whereas NOTCH3 in patients was previously mainly associated with the CADASIL syndrome, our observations in the mouse and a human cohort support a novel role in congenital heart defects and laterality defects.
Heterotaxy (1/13,000 birth) encompasses a heterogenous spectrum of defects. To untangle the developmental trajectories, we performed the first longitudinal analysis, an experimental tour de force imaging the same individual at two stages. We show that the shape of heart looping only partially predicts congenital heart defects. Whereas the position of ventricles was considered to be fixed during heart looping, we uncover an additional fetal step able to modulate ventricle position in heterotaxy. The discovery in the mouse of revertants, which underwent leftward embryonic heart looping but end in a D-LOOP configuration at birth has clinical impact. It questions the nomenclature of D-LOOP/L-LOOP in the light of developmental mechanisms. In patients, the structural ventricle anomalies observed in mouse revertants are associated with poorer prognosis.
