Médecine sensorielle de précision : des mécanismes des maladies aux solutions thérapeutiques
L’ouïe, l’équilibre et la vision sont essentiels à notre manière de percevoir, d’interpréter et d’interagir avec le monde. Pourtant, les troubles sensoriels — souvent invisibles et progressifs — touchent des millions de personnes, affectant profondément leur qualité de vie. À l’échelle mondiale, plus de 460 millions de personnes vivent avec une perte auditive invalidante, un chiffre qui devrait dépasser un milliard d’ici 2050. La dysfonction vestibulaire concerne entre 403 millions et 725 millions d’individus, soit jusqu’à 9 % de la population mondiale.
Parallèlement, plus de 285 millions de personnes souffrent d’une déficience visuelle sévère. Le poids économique et sociétal est colossal, soulignant un besoin urgent d’innovations scientifiques et médicales.
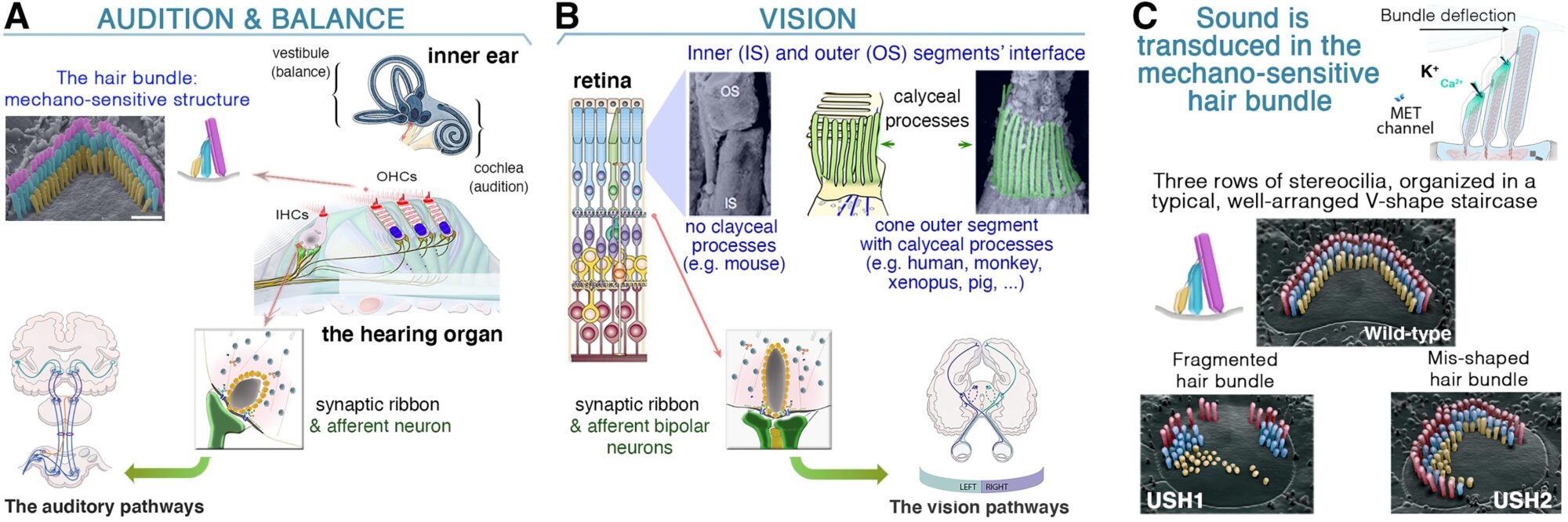
(A, B) Similarités entre l’œil et l’oreille interne. L’audition dépend du traitement des ondes sonores au niveau des touffes ciliaires qui couronnent les cellules ciliées internes (CCI) et externes (CCE) dans la cochlée (A). Les signaux lumineux sont transduits par les segments externes des photorécepteurs dans la rétine (B). Dans les cellules ciliées (A) et les photorécepteurs (B), le neurotransmetteur (glutamate) est libéré dans les zones actives synaptiques, où les vésicules synaptiques sont ancrées à une structure dense aux électrons appelée « ruban synaptique ». (C) L’architecture de la touffe ciliaire (de type sauvage) est altérée différemment en l’absence des protéines Usher de types 1 et 2.
Une approche intégrative centrée sur les déficiences sensorielles progressives
Au sein de l’unité Déficits Sensoriels Progressifs (DSP) à l’Institut de l’Audition, Institut reConnect, centre de l’Institut Pasteur, notre mission est de repousser les frontières des neurosciences sensorielles et des thérapies associées.
Nos recherches s’articulent autour de la complexité des troubles affectant l’oreille interne (audition et équilibre) et la rétine (vision), avec un intérêt particulier pour les pathologies progressives, souvent postnatales et aggravées par l’âge ou l’environnement. Contrairement à d’autres espèces, chez les mammifères, de nombreux processus façonnant le capital auditif se déroulent avant la naissance, déterminant une vulnérabilité particulière au cours de la vie.
De la génétique à la thérapie : comprendre, expliquer, traiter
Notre approche consiste à déconstruire le réseau complexe des causes des déficits sensoriels en nous concentrant sur les formes héréditaires, comme celles observées dans le syndrome d’Usher. Nous étudions les atteintes héréditaires tardives et progressives, en tant que modèles de vieillissement accéléré, dans le but de mieux comprendre les mécanismes sous-jacents aux troubles sensoriels plus fréquents, tels que la presbyacousie ou la perte liée au bruit. Nos objectifs s’articulent autour de trois axes complémentaires :
- Comprendre l’origine des déficiences,
- Expliquer la variabilité des symptômes,
- Traiter de manière personnalisée.
En pratique, ils convergent vers une ambition unique : mieux comprendre pour mieux soigner.
De l’observation clinique aux modèles animaux
En partant des observations cliniques, nous développons des modèles animaux spécifiques, capables de reproduire la diversité des trajectoires pathologiques humaines.
Ces modèles — notamment murins et porcins — font l’objet d’analyses multiscalaires intégrant la génétique, la biologie moléculaire, l’imagerie, l’électrophysiologie et le comportement. Notre objectif est de déterminer le quoi, le où, le quand et le comment du développement des déficits sensoriels, en suivant leur progression dans des contextes standardisés ou perturbés, avec ou sans intervention thérapeutique.
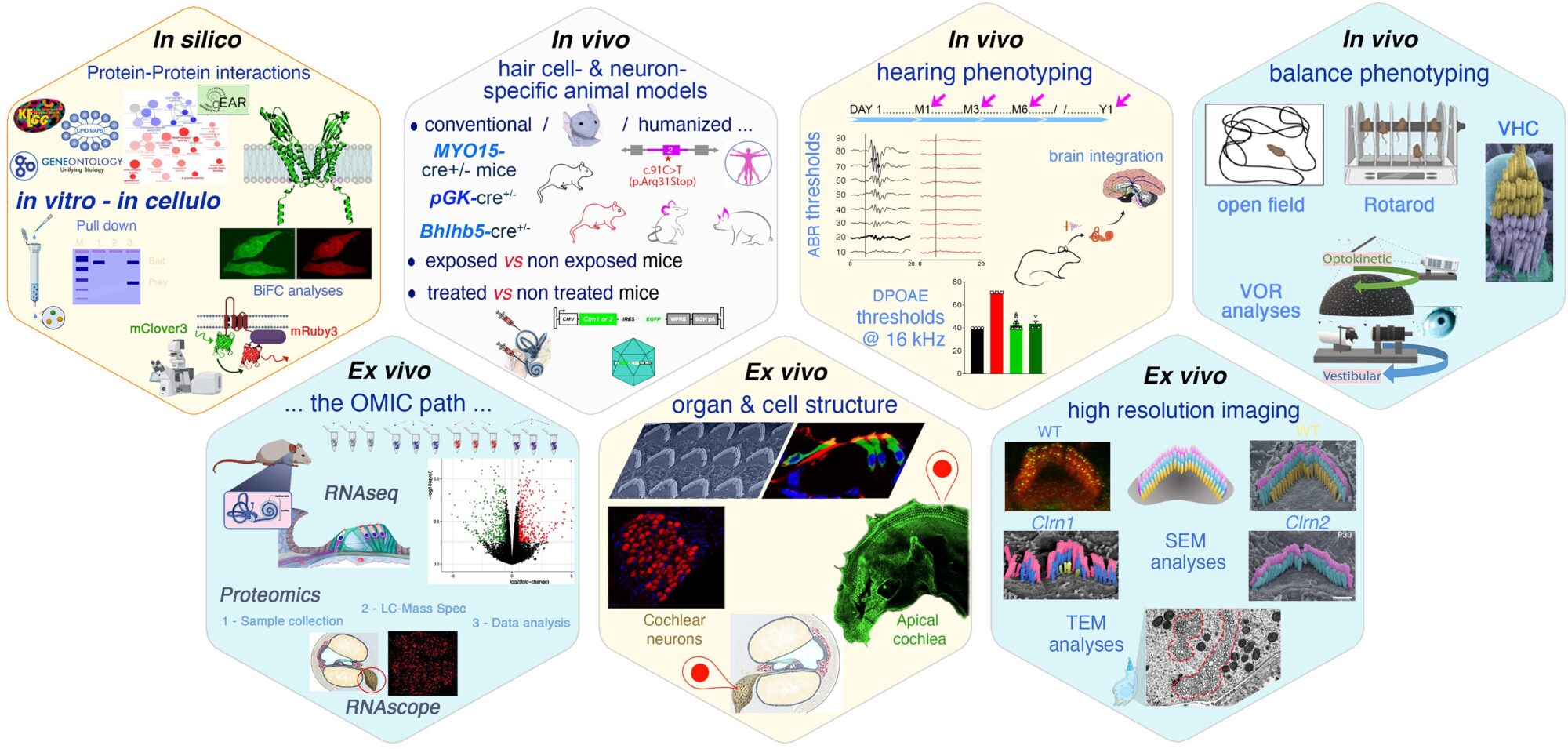
Un pipeline de phénotypage innovant
Notre pipeline de phénotypage approfondi permet une cartographie précise des altérations fonctionnelles et structurelles des organes sensoriels. Grâce à une approche intégrative, nous pouvons suivre la sensibilité auditive, mais aussi les déficits vestibulaires et visuels, à toutes les étapes de la maladie.
Ce workflow nous permet d’identifier les perturbations précoces avant qu’elles ne deviennent irréversibles, et de tester l’effet de différentes interventions, y compris les thérapies géniques.
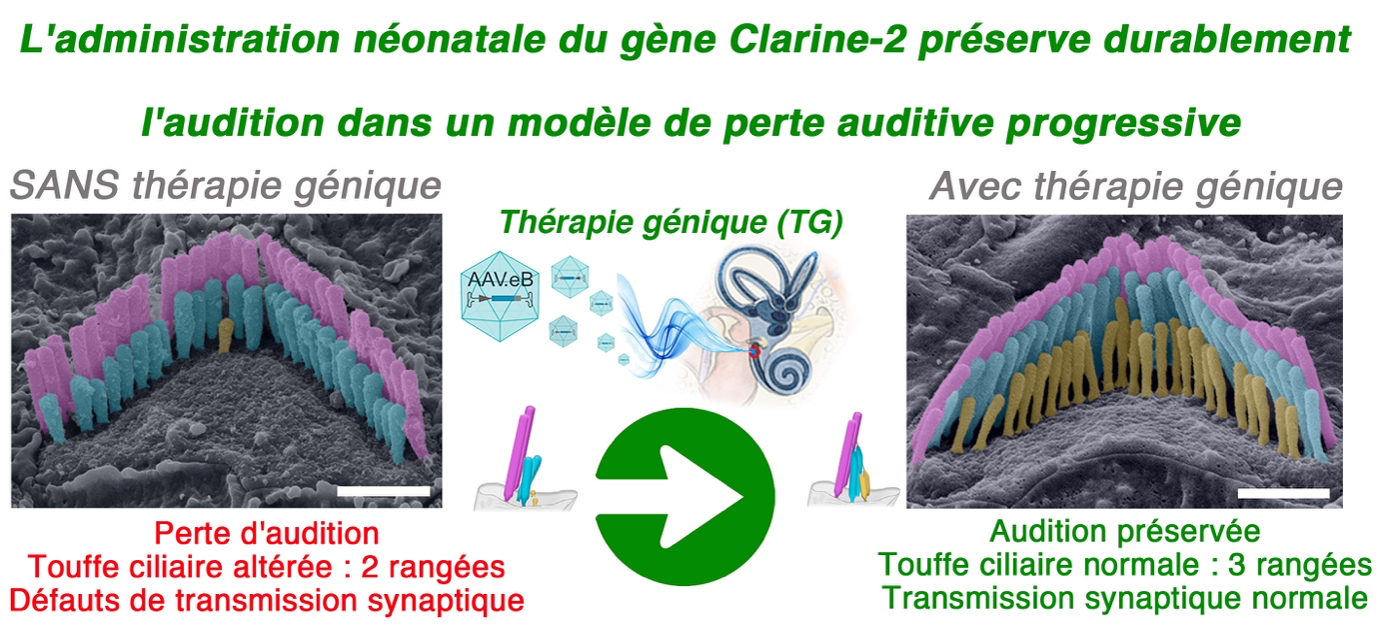
Modèles et solutions thérapeutiques
Nous explorons des stratégies thérapeutiques ciblées, adaptées à la diversité des mutations génétiques impliquées dans les déficiences auditives et visuelles. Par exemple, dans certains modèles (voir le cas du gène Clrn2 ci-dessous), une thérapie de remplacement génique précoce permet de préserver l’audition normale et d’éviter la dégénérescence des cellules ciliées sensorielles. Nos modèles servent également de plateforme pour valider la pathogénicité de mutations humaines et comprendre l’impact des variations évolutives sur la fonction des gènes.