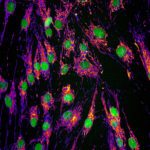

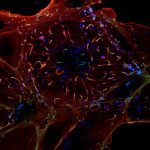




Genetic mutations in the gene Optic Atrophy 1 (OPA1) cause autosomal dominant optic atrophy (DOA), one of the most common forms of mitochondrial disease where mitochondria are fragmented in small vesicles. While most of patients develop isolated deterioration of the optic nerve, about 20% of them develop a more severe neurological disease, called DOA+, that cannot be fully explained by the disease-causing mutation in OPA1.
Combining image analysis using supervised machine learning to quantify mitochondrial morphology defects and high-throughput genetic screening of 1,531 nuclear-encoded mitochondrial genes (called the mitome), Tim Wai and his colleagues analysed fibroblasts from both healthy individuals and patients suffering from DOA+. They then identified genes involved in mitochondrial morphology maintenance and fragmentation, among which PGS1, a key enzyme in cardiolipin biosynthesis. PGS1 depletion reduces mitochondrial fission and restores normal mitochondrial morphology to OPA1-deficient fibroblasts, but does not recover all defects displayed by the mitochondria. This study shows mitochondrial defects linked to OPA1 deficiency in DOA patients are also modulated by the action of other mitochondrial genes in a more intricate fashion, with clinical implications for diagnosis.
Source
High-throughput screening identifies suppressors of mitochondrial fragmentation in OPA1 fibroblasts. EMBO Molecular Medicine, 20 May 2021.
To read the article: here
To read the comment published in EMBO Mol Med: here
To read the press release: here