Widespread macromolecular interaction perturbations in human genetic disorders
Scientific Fields
Diseases
Organisms
Applications
Technique
Published in Cell - 23 Apr 2015
Sahni N, Yi S, Taipale M, Fuxman Bass JI, Coulombe-Huntington J, Yang F, Peng J, Weile J, Karras GI, Wang Y, Kovács IA, Kamburov A, Krykbaeva I, Lam MH, Tucker G, Khurana V, Sharma A, Liu YY, Yachie N, Zhong Q, Shen Y, Palagi A, San-Miguel A, Fan C, Balcha D, Dricot A, Jordan DM, Walsh JM, Shah AA, Yang X, Stoyanova AK, Leighton A, Calderwood MA, Jacob Y, Cusick ME, Salehi-Ashtiani K, Whitesell LJ, Sunyaev S, Berger B, Barabási AL, Charloteaux B, Hill DE, Hao T, Roth FP, Xia Y, Walhout AJ, Lindquist S, Vidal M
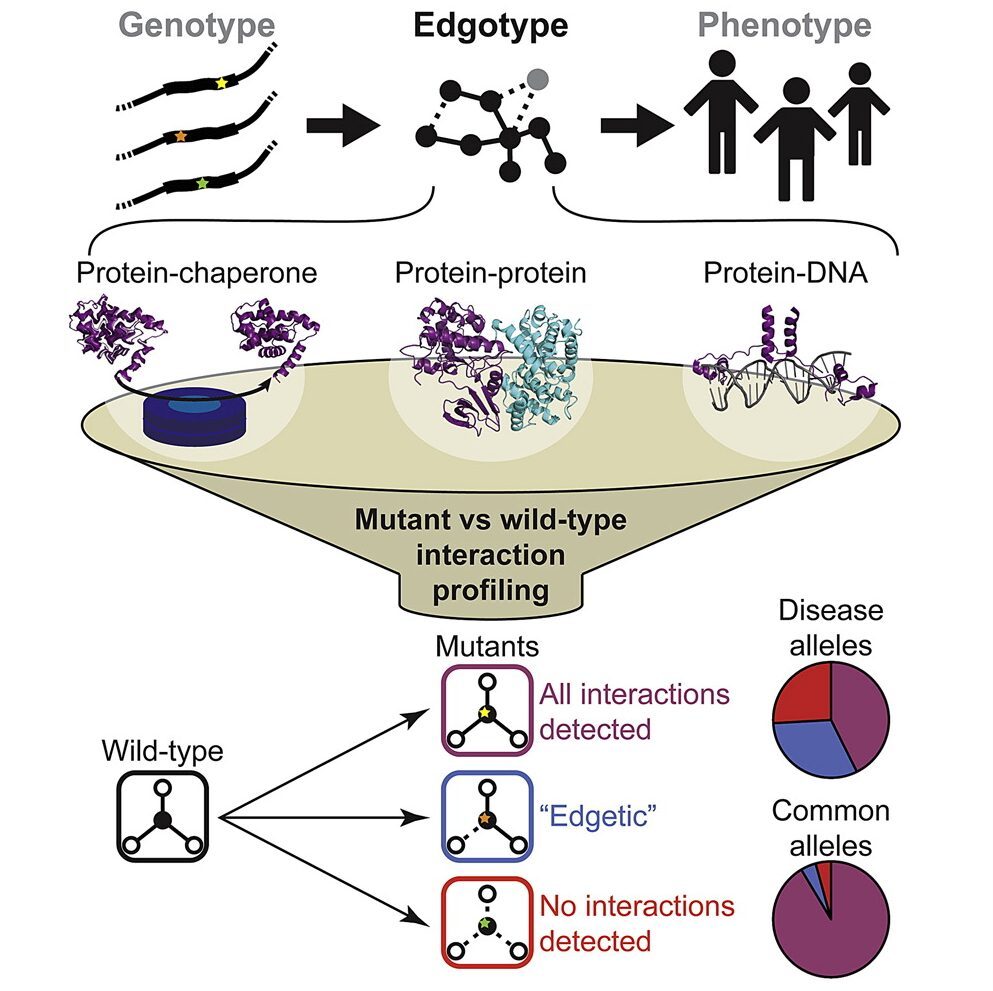
How disease-associated mutations impair protein activities in the context of biological networks remains mostly undetermined. Although a few renowned alleles are well characterized, functional information is missing for over 100,000 disease-associated variants. Here we functionally profile several thousand missense mutations across a spectrum of Mendelian disorders using various interaction assays. The majority of disease-associated alleles exhibit wild-type chaperone binding profiles, suggesting they preserve protein folding or stability. While common variants from healthy individuals rarely affect interactions, two-thirds of disease-associated alleles perturb protein-protein interactions, with half corresponding to “edgetic” alleles affecting only a subset of interactions while leaving most other interactions unperturbed. With transcription factors, many alleles that leave protein-protein interactions intact affect DNA binding. Different mutations in the same gene leading to different interaction profiles often result in distinct disease phenotypes. Thus disease-associated alleles that perturb distinct protein activities rather than grossly affecting folding and stability are relatively widespread.